
INVESTIGATORS
About AIM HIGHer
The AIM HIGHer Clinical Trial is a prospective, multicenter, randomized, quadruple-blind, sham-controlled, two-part embedded trial of the safety and efficacy of CCM® therapy delivered by the Optimizer® Smart Mini in subjects with heart failure and an LVEF ≥40% and ≤60%. Subjects will be enrolled at approximately 150 sites in the US.
All subjects will undergo screening and baseline testing. All eligible subjects will be implanted with the Optimizer system.
Subjects will be randomized in a 2:1 ratio to either CCM therapy ON (CCM therapy group) or CCM therapy OFF (Sham group). The trial will be blinded to the treatment assignment of the device for 18-months. Subjects in the Sham group will have CCM therapy turned ON after completing the 18-month study visit. Subjects enrolled during Part I (450 subjects) of the trial will continue follow-up through the end of Part II (up to an additional 1,050 subjects) and contribute data to both parts of the trial. Each part of the trial is distinguished by a separate scientific purpose. The specific purpose of each part is:
Part I – Establish safety and effectiveness based on functional capacity and health status.
Part II – Establish safety and effectiveness based on clinical outcome data.
Part I

Part I - Primary Efficacy Objectives
- Change in 6-minute walk distance (6MWD) from baseline to 6 months. The 6MW test is a non-invasive exercise test used to evaluate functional capacity in which the distance walked on a flat surface in a six- minute time period is measured, in accordance with the 6MWT Instructions and Documentation detailed in the study procedures (to be provided separately).

Part I - Primary Safety Objective
- The change in the health status from baseline to 6 months as assessed by the KCCQ CSS. The KCCQ is a 23-item patient-reported outcome instrument with a total possible score of 0-100; the higher the score indicates better health-related quality of life. The CSS is a 13-item section within KCCQ that includes the domains that measure symptoms (frequency and burden) and physical limitations associated with heart failure. A licensing agreement will be obtained from Outcomes Instruments, LLC prior to study initiation.
Part II
Part II – Primary Objective
The hierarchical composite of cardiovascular-related mortality, heart failure hospitalization, and urgent heart failure visits requiring IV treatment at 18-months and change in KCCQ CSS from baseline at 12-months. The CEC will review, adjudicate and classify all reported deaths, hospitalizations, and urgent health care visits through the 18-month blinded phase of the trial. The CEC will adjudicate each death as attributable to cardiovascular causes or not, will classify each episode of care in the hospital setting as attributable primarily to heart failure or not, and classify urgent health care visits as attributable to heart failure requiring the use of IV therapy or not. In addition, the KCCQ CSS will be assessed as previously described in the Part 1 primary efficacy endpoint.
Individuals must meet all the following criteria:
1. Signed and dated informed consent form;
2. Male or non-pregnant female, 18 years or older;
3. Diagnosed with symptomatic heart failure;
4. LVEF ≥40 and <60% (as assessed by site echo)
5.
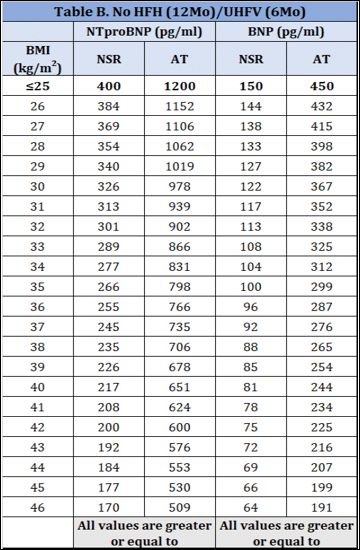
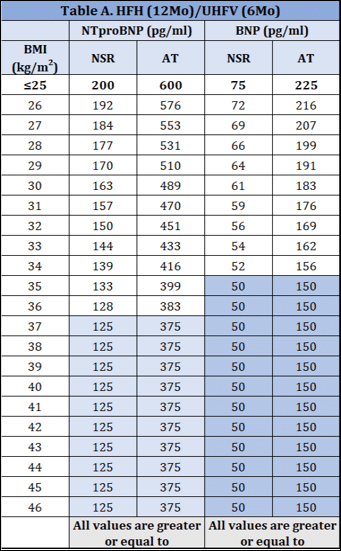
A. Heart failure hospitalization within 12 months prior to study consent OR an urgent heart failure visit requiring IV therapy within 6 months prior to study consent, AND elevated BMI-adjusted natriuretic peptide values
OR
B. If there is no heart failure hospitalization within 12 months prior to study consent OR an urgent heart failure visit requiring IV therapy within 6 months prior to study consent, an elevated BMI-adjusted natriuretic peptide value must be achieved
6. Subjects must meet one of the following conditions:
• Have stable, scheduled oral loop diuretic treatment (not just PRN) for a minimum of 30 days before providing study consent unless there is a documented allergy or intolerance.
• Eligibility for enrollment is maintained for patients on an SGLT2 inhibitor without prescribed concurrent standing loop diuretic therapy if investigators provide instructions for a flexible PRN diuretic regimen (deemed appropriate by the clinician in response to symptoms or weight gain).
Note: Stable is defined as no more than a 100% increase or 50% decrease in dose within the last 30 days. A one-time hold of diuretic dosing for 24 hours during the 30-day period is allowed and not an exclusionary event.
An individual who meets any of the following criteria will be excluded from participation in this study:
1. Resting ventricular rate <50 or >110 bpm;
2. Resting systolic blood pressure <100 or >160 mmHg;
3. BMI greater than 46
4. Any severe valvular stenotic disease or any severe valvular regurgitation;
5. Mechanical tricuspid valve;
6. Complex congenital heart disease (e.g. atrial septal defects < 1cm are acceptable);
7. Exercise tolerance limited by a condition other than heart failure (e.g., angina, significant pulmonary disease, or currently requires home oxygen or oral steroids, peripheral vascular disease, orthopedic or rheumatologic conditions) that, in the opinion of the investigator, contributes significantly to the primary symptoms of shortness of breath and/or exercise intolerance;
8. Unable to walk at least 100 meters or walks more than 450 meters during a 6MWT;
9. A KCCQ CSS score higher than 85;
10. Hypertrophic, infiltrative/restrictive (e.g., amyloidosis, sarcoidosis, hemochromatosis, Fabry, or radiation-induced heart disease), or inflammatory cardiomyopathy as documented in the medical record; Note: Refer to Section 9.4.1. Screening Evaluation and Baseline Visit for guidance on amyloidosis screening.
11. Unstable angina pectoris within 30 days prior to study consent;
12. Acute, decompensated heart failure requiring IV therapy or ultrafiltration within 30 days prior to baseline echo.
13. Receiving cardiac resynchronization therapy (CRT);
14. Scheduled for a cardiac surgery or a percutaneous cardiac intervention (PCI) or have undergone cardiac surgery within 90 days or a PCI procedure within 30 days prior to study consent;
15. Myocardial infarction within 90 days prior to study consent;
16. Prior heart transplant or current ventricular assist device;
17. Planning to become pregnant during the study;
18. Dialysis (permanent) or GFR <20 ml/min/1.73m2;
19. Participating in another investigational study;
Note: Patients may be participating in registry/observational studies.
20. Currently undergoing active chemotherapeutic and/or radiation treatment for cancer or has a history of chemotherapy during the 2-year period prior to study consent;
21. Expected lifespan of less than 18 months from time of study consent;
22. Unable to follow through study protocol for any reasons (social support, distance from research center, insurance denial, mental illness etc.) in the investigator’s judgement.
Note: Concomitant devices involving intracardiac leads, if required, should be implanted a minimum of either 31 days prior to or 31 days after the Optimizer implant procedure.
© 2024 Impulse Dynamics Terms of Use | Data Protection | Vulnerability Disclosure | Terms and Conditions